Aplastic Anemia
Content of This Page
1- Introduction
2- Pathophysiology
3- Clinical Features
4- Stages of The Disease
5- Investigations
6- Treatment
7- Prognosis & Follow-up
8- What Should You Avoid
Introduction
Aplastic anaemia is a rare but potentially life-threatening condition caused by failure of the bone marrow to produce blood cells, leading to pancytopenia—a reduction in red cells, white cells, and platelets. It may be acquired or inherited, and the majority of cases are idiopathic.The condition often presents with a combination of anaemia (fatigue), neutropenia (infections), and thrombocytopenia (bleeding). Prompt diagnosis and treatment are essential to avoid fatal complications.
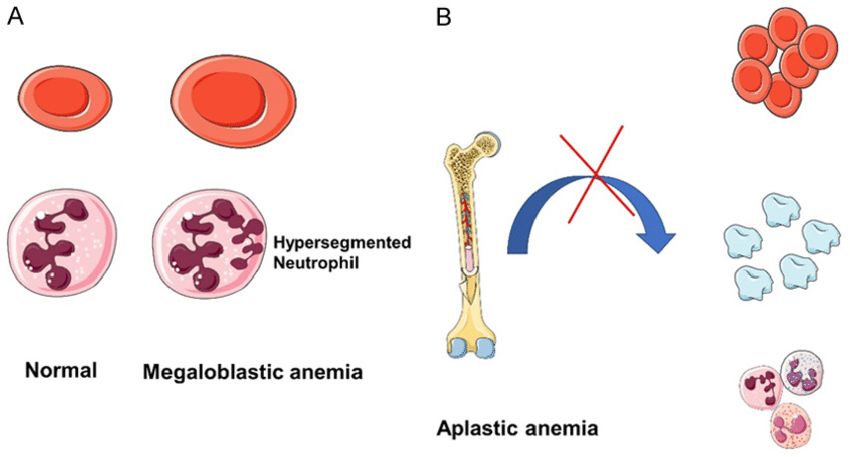
Pathophysiology
1. Loss or Suppression of Haematopoietic Stem Cells
The primary defect in aplastic anaemia lies in the bone marrow stem cells responsible for producing:
Erythrocytes (RBCs) → leading to anaemia
Leukocytes (WBCs) → leading to neutropenia and infections
Platelets → leading to thrombocytopenia and bleeding
2. Causes of Stem Cell Suppression
a. Immune-Mediated Destruction (Most Common in Acquired Cases)
Autoreactive T cells target and destroy haematopoietic stem cells.
Suggested by clinical response to immunosuppressive therapy (e.g. anti-thymocyte globulin and ciclosporin).
b. Direct Toxic Damage
Exposure to drugs, chemicals (e.g. benzene), or ionising radiation can directly damage stem cells.
c. Viral Infections
Particularly hepatitis viruses, parvovirus B19, EBV, and HIV—may lead to immune or direct cytotoxic suppression of marrow.
d. Inherited Genetic Defects
Rare causes like Fanconi anaemia or Dyskeratosis congenita involve defects in DNA repair or telomerase function.
3. Hypocellular Marrow
Bone marrow becomes fatty and hypocellular, with marked reduction in haematopoietic precursors.
This leads to inadequate production of all blood cell lines (pancytopenia).
Symptoms
| Cytopenia Type | Symptom Cluster |
|---|---|
| Anaemia | Fatigue, pallor, dyspnoea, palpitations |
| Neutropenia | Infections, sore throat, ulcers, fever |
| Thrombocytopenia | Bruising, petechiae, bleeding gums/nose |
Stages of The Disease
1. Non-Severe Aplastic Anaemia (NSAA)
Hypocellular bone marrow
Blood counts are reduced but do not meet criteria for severe disease.
Patients may be asymptomatic or have mild symptoms.
2. Severe Aplastic Anaemia (SAA)
(Camitta Criteria – all must be met):
Bone marrow cellularity < 25%
or
25–50% with <30% haematopoietic cells
AND at least two of the following:Neutrophils < 0.5 × 10⁹/L
Platelets < 20 × 10⁹/L
Reticulocytes < 20 × 10⁹/L
3. Very Severe Aplastic Anaemia (VSAA)
Same criteria as SAA, plus:
Neutrophils < 0.2 × 10⁹/L
This group is at highest risk of severe infections and requires urgent treatment.
Investigations
1. Initial Blood Tests
| Test | Expected Finding |
|---|---|
| Full blood count (FBC) | Pancytopenia: ↓Hb, ↓WBC, ↓platelets |
| Reticulocyte count | Low, indicating reduced red cell production |
| Peripheral blood film | Normocytic, normochromic anaemia; no blasts |
| Liver and renal function | To exclude systemic causes |
| Vitamin B12/folate | To exclude megaloblastic anaemia |
| Serum ferritin and iron | Typically normal or elevated (due to transfusions) |
| ESR/CRP | Usually normal (helps rule out inflammatory conditions) |
2. Bone Marrow Examination
a. Bone Marrow Aspiration and Trephine Biopsy
Key diagnostic test
Findings:
Hypocellular marrow (fatty replacement)
Reduced or absent myeloid, erythroid, and megakaryocyte precursors
No fibrosis or abnormal infiltrates
Helps distinguish aplastic anaemia from leukaemia, lymphoma, and myelofibrosis.
3. Exclusion of Other Causes
| Test | Rationale |
|---|---|
| Viral serologies (Hepatitis B/C, EBV, HIV, parvovirus B19) | Rule out viral-induced marrow suppression |
| Autoimmune screening (ANA, dsDNA) | If autoimmune aetiology suspected |
| Cytogenetics / FISH | To exclude myelodysplastic syndromes |
| Flow cytometry (PNH clone test) | Rule out paroxysmal nocturnal haemoglobinuria |
| Chromosome breakage test | In suspected Fanconi anaemia (inherited cases) |
Treatment
1. Supportive Care
(Given in all stages while awaiting definitive therapy)
| Supportive Measure | Purpose |
|---|---|
| Red cell transfusions | For symptomatic anaemia |
| Platelet transfusions | For active bleeding or counts <10 × 10⁹/L |
| Antimicrobial therapy | For neutropenic infections |
| Infection prophylaxis | Neutropenic precautions ± antifungals |
| Growth factors (e.g. G-CSF) | Sometimes used in neutropenic episodes |
2. Definitive Treatment
a. Allogeneic Stem Cell Transplantation (SCT)
First-line for patients <40 years with a matched sibling donor.
Offers the only potential cure.
Requires good performance status and no severe infections.
b. Immunosuppressive Therapy (IST)
First-line for:
Older patients (>40),
Those without a matched donor,
Or as second-line if transplant fails.
Regimen:
Anti-thymocyte globulin (ATG) + Ciclosporin (CSA)
± Eltrombopag (a thrombopoietin receptor agonist that may improve response)Response may take weeks to months; regular monitoring is essential.
3. Treatment by Severity
| Severity | First-line Treatment |
|---|---|
| Very severe (VSAA) | SCT (if young + matched donor) or ATG + CSA |
| Severe (SAA) | Same as above |
| Non-severe (NSAA) | Observation or IST if symptomatic or progressive cytopenias |
4. Monitoring & Follow-up
Blood counts regularly (weekly → monthly)
Monitor for relapse or clonal evolution (e.g. MDS, AML)
Long-term complications of immunosuppression (infections, secondary malignancies)
Prognosis & Follow-up
1. Prognosis
| Factor | Impact on Prognosis |
|---|---|
| Age | Better outcomes in younger patients (<40 years) |
| Severity of cytopenias | Very severe aplasia carries highest risk of mortality |
| Treatment response | Good IST/SCT response = higher survival |
| Stem cell transplantation | Offers potential cure (5-year survival > 80%) |
| Complications | Infections, bleeding, iron overload from transfusions |
Long-Term Outcomes
Survival >75–90% with successful treatment
Risk of relapse, especially after IST, occurs in ~30% of patients
Some may develop clonal evolution:
Myelodysplastic syndrome (MDS)
Acute myeloid leukaemia (AML)
Paroxysmal nocturnal haemoglobinuria (PNH)
2. Follow-Up Care
a. Clinical Monitoring
Regular symptom review: fatigue, bleeding, infections
b. Blood Count Monitoring
Initially weekly, then monthly once stable
Watch for trends suggesting relapse or evolution
c. Post-IST Monitoring
Renal function (due to ciclosporin)
Drug levels (if applicable)
Tapering immunosuppressives cautiously over 6–12 months
d. Post-SCT Monitoring
Graft function (chimerism)
Graft-versus-host disease (GVHD)
Opportunistic infection surveillance
e. Other Surveillance
Iron overload from transfusions (consider ferritin + MRI)
Screening for clonal disorders (e.g. periodic PNH clone test, cytogenetics)
What Should You Avoid
| What to Avoid | Why |
|---|---|
| Myelotoxic drugs | Risk of worsening pancytopenia |
| Live vaccines (during IST) | May cause disseminated infection |
| Invasive procedures without cover | Risk of bleeding in thrombocytopenia |
| Crowded/public places | Neutropenia = high infection risk |
| Unscreened blood products | Risk of alloimmunisation and transfusion reaction |
| Uncooked foods/gardening | Fungal or bacterial infection risk |