Myelodysplastic Syndromes (MDS)
Content of This Page
1- Introduction
2- Pathophysiology
3- Classification
4- Clinical Features
5- Investigations
6- Treatment
7- Prognosis & Follow-up
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal haematopoietic stem cell disorders characterised by ineffective blood cell production (dysplasia) and a risk of progression to acute myeloid leukaemia (AML). MDS is most commonly seen in older adults, and presents with cytopenias (especially anaemia), often found incidentally on blood tests. The hallmark of MDS is a mismatch between a hypercellular bone marrow and peripheral cytopenias, due to premature apoptosis of abnormal marrow precursors.
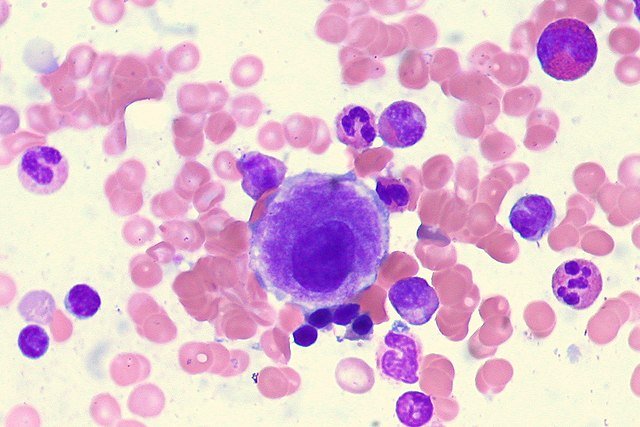
Pathophysiology
1. Clonal Haematopoietic Stem Cell Mutation
MDS originates from somatic mutations in a single haematopoietic stem cell, which clonally expands.
Common genetic mutations include those in:
Spliceosome genes (e.g. SF3B1)
Epigenetic regulators (e.g. TET2, DNMT3A)
TP53 (associated with poor prognosis)
2. Ineffective Haematopoiesis
The mutated stem cells retain the ability to differentiate, but the process is inefficient and dysplastic.
Results in:
Hypercellular bone marrow (cells are produced but undergo apoptosis)
Peripheral blood cytopenias (due to early cell death before maturation)
3. Cytopenias and Dysplasia
All three blood cell lines may be affected:
Red cells → anaemia (often macrocytic)
White cells → neutropenia, ↑ infection risk
Platelets → thrombocytopenia, bleeding
Dysplastic features on blood film or marrow:
Pseudo–Pelger-Huet neutrophils
Micromegakaryocytes
Ring sideroblasts (especially in SF3B1 mutation)
4. Progression to AML
Over time, additional mutations may accumulate, especially in genes like TP53 or RUNX1, leading to:
Loss of differentiation capacity
Blast cell expansion
Transformation into acute myeloid leukaemia (AML)
Risk of progression varies by cytogenetics and blast count at diagnosis.
Classification
1. WHO Classification (2022 Update – Simplified)
The WHO classification is based on:
Blast percentage in bone marrow/peripheral blood
Presence of dysplasia
Cytogenetic abnormalities
Ring sideroblasts
Molecular mutations
Major WHO Subtypes
| Subtype | Key Features |
|---|---|
| MDS with single lineage dysplasia (MDS-SLD) | Dysplasia in one cell line; <5% blasts; no ring sideroblasts |
| MDS with multilineage dysplasia (MDS-MLD) | Dysplasia in ≥2 lineages; <5% blasts |
| MDS with excess blasts (MDS-EB1/EB2) | 5–9% blasts (EB1), or 10–19% (EB2) → high risk of progression to AML |
| MDS with isolated del(5q) | Anaemia, normal or ↑ platelet count; good prognosis |
| MDS with ring sideroblasts (MDS-RS) | ≥15% ring sideroblasts (or ≥5% if SF3B1 mutation present) |
| MDS, unclassifiable (MDS-U) | Does not fit other categories; rare |
2. IPSS-R (Revised International Prognostic Scoring System)
Used to stratify prognosis and guide treatment. Based on:
Cytogenetic risk category
Bone marrow blast percentage
Haemoglobin level
Platelet count
Absolute neutrophil count
Risk Categories:
| IPSS-R Score | Risk Group | Estimated Median Survival |
|---|---|---|
| 0–1.5 | Very low | >8 years |
| >1.5–3.0 | Low | ~5 years |
| >3.0–4.5 | Intermediate | ~3 years |
| >4.5–6.0 | High | ~1.5 years |
| >6.0 | Very high | <1 year |
3. By Etiology
| Type | Description |
|---|---|
| Primary MDS | De novo, no identifiable cause (most common) |
| Secondary MDS | Related to previous chemotherapy, radiotherapy, or toxins |
Secondary MDS often has poorer prognosis and complex cytogenetics.
Clinical Features
| Cell Line Affected | Symptoms/Signs |
|---|---|
| Red cells (anaemia) | Fatigue, pallor, breathlessness |
| White cells (neutropenia) | Infections, mouth ulcers, fever |
| Platelets (thrombocytopenia) | Bleeding, petechiae, easy bruising |
Investigations
| Investigation Area | Key Tests |
|---|---|
| Cytopenia detection | FBC, reticulocyte count |
| Morphological dysplasia | Blood film, marrow aspirate & biopsy |
| Confirm clonality | Cytogenetics, molecular markers |
| Rule out mimics | B12, folate, thyroid function, viral serologies |
Treatment
| MDS Risk Group | Treatment Approach |
|---|---|
| Low-risk (IPSS-R low/very low) | Supportive care ± ESAs, luspatercept, lenalidomide (if del(5q)) |
| Intermediate-risk | May consider hypomethylating agents or SCT if younger |
| High/very high-risk | Azacitidine or decitabine ± SCT if eligible |
| All patients | Transfusions, infection control, iron chelation if needed |
Prognosis & Follow-up
| Follow-Up Focus | Details |
|---|---|
| Prognosis tool | IPSS-R (risk stratification) |
| Monitoring intervals | FBC every 1–3 months; marrow if deterioration |
| Common complications | AML progression, iron overload, infections |
| Endpoints of follow-up | Stability, response to treatment, or transformation |