Neurofibromatosis Type I (NF1)
Content of This Page
1- Introduction
2- Causes
3- Symptoms
4- Investigations & Lab Results
5- Complications
6- Treatment
Introduction
Neurofibromatosis Type I (NF1), also known as von Recklinghausen disease, is a common autosomal dominant genetic disorder that primarily affects the nervous system, skin, and eyes. It is characterized by the development of multiple benign nerve sheath tumors (neurofibromas) and distinctive skin findings, including café-au-lait spots and axillary/inguinal freckling.
NF1 is caused by mutations in the NF1 gene located on chromosome 17, which encodes the neurofibromin protein, a tumor suppressor that regulates cell growth. Loss of neurofibromin function leads to uncontrolled cell proliferation and tumor formation.
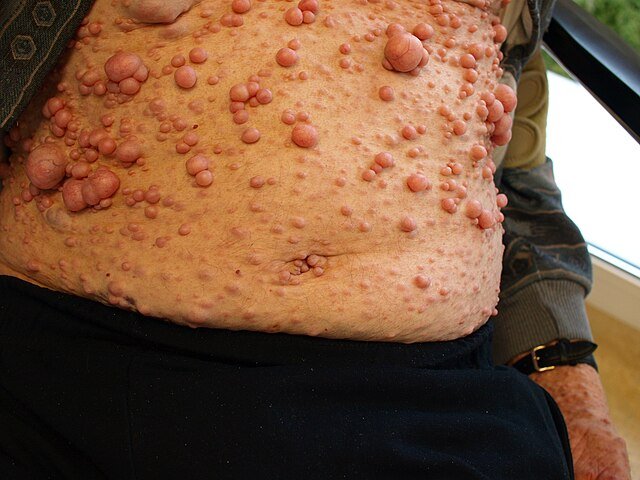
Causes
1. Genetic Cause
NF1 gene mutation:
This gene encodes neurofibromin, a tumor suppressor protein that helps regulate cell growth by inhibiting the RAS signaling pathway.
A mutation leads to loss of neurofibromin function, resulting in uncontrolled cell division and tumor formation, especially in nerve tissue.
2. Inheritance Pattern
Autosomal Dominant Inheritance:
Each child of an affected parent has a 50% chance of inheriting the condition.
The disease shows variable expressivity, meaning symptoms can vary widely even among family members with the same mutation.
3. Sporadic Mutations
Approximately 50% of cases are due to new (de novo) mutations, meaning they occur spontaneously without a family history.
These mutations happen during early embryonic development or in the germ cells of one parent.

Symptoms
1 . Dermatologic Features
Café-au-lait spots
Flat, light-brown skin patches
Usually >6 lesions >5 mm in prepubertal children or >15 mm in postpubertal individuals
Axillary or inguinal freckling
Clusters of freckles in armpits or groin (Crowe’s sign)
Cutaneous neurofibromas
Soft, benign tumors on or under the skin
May increase in number and size with age
Plexiform neurofibromas
Larger, deeper tumors along nerves
Often congenital and may cause disfigurement or compression of nearby structures
2. Neurological Features
Learning disabilities (seen in ~50% of children)
Often mild but may affect academic performance
Attention-deficit/hyperactivity disorder (ADHD)
Seizures (less common)
Headaches
3. Ophthalmologic Features
Lisch nodules
Benign pigmented iris hamartomas
Asymptomatic and detectable on slit-lamp examination
Optic pathway gliomas
Tumors of the optic nerve
May cause vision loss or proptosis (bulging eye)
4. Skeletal Abnormalities
Scoliosis (curvature of the spine)
Long bone dysplasia
Especially tibial bowing or pseudarthrosis (non-healing fracture)
Short stature
5. Other Possible Symptoms
Hypertension (can result from renal artery stenosis or pheochromocytoma)
Macrocephaly (large head size)
Delayed or early puberty
Investigations & Lab Results
1. Clinical Diagnosis
Diagnosis is based on the National Institutes of Health (NIH) criteria requiring two or more of the following:
Six or more café-au-lait spots (≥5 mm in children, ≥15 mm in adults)
Two or more neurofibromas or one plexiform neurofibroma
Axillary or inguinal freckling
Optic glioma
Two or more Lisch nodules (iris hamartomas)
A distinctive bone lesion (e.g., sphenoid dysplasia, tibial pseudarthrosis)
A first-degree relative with NF1
2. Imaging Studies
MRI of the brain and spine:
To detect optic pathway gliomas, plexiform neurofibromas, and other CNS tumors
Assess for skeletal abnormalities or spinal cord involvement
X-rays:
To evaluate bone dysplasia, scoliosis, or pseudarthrosis
Ultrasound or CT scan:
If visceral neurofibromas or vascular abnormalities are suspected
3. Ophthalmologic Examination
Slit-lamp exam:
To identify Lisch nodules (pigmented iris hamartomas)
Visual field testing and fundus exam:
To assess optic nerve function, especially if optic glioma is suspected
4. Genetic Testing
NF1 gene mutation analysis:
Useful in unclear or early cases, prenatal diagnosis, or family counseling
Can confirm diagnosis but not always necessary due to clinical criteria
5. Other Laboratory Tests
Generally not required for diagnosis
May be done to evaluate associated conditions (e.g., hypertension workup, endocrine evaluation)
Complications
1. Tumor-related Complications
Malignant Peripheral Nerve Sheath Tumors (MPNST):
Malignant transformation of plexiform neurofibromas (rare but serious)
Presents with rapid growth, pain, or neurological deficits
Plexiform neurofibromas:
Can cause disfigurement, pain, and functional impairment
Difficult to completely remove surgically
Optic pathway gliomas:
May cause vision loss or blindness if untreated
2. Neurological Complications
Seizures (rare)
Headaches and migraines
Cognitive impairment and learning disabilities
Developmental delays and behavioral problems
3. Skeletal Complications
Scoliosis:
May progress and cause deformity or respiratory problems
Tibial dysplasia and pseudarthrosis:
Leads to bone fragility, fractures, and non-healing wounds
Other bone abnormalities:
Sphenoid wing dysplasia causing orbital defects
Short stature or macrocephaly
4. Vascular Complications
Hypertension:
Due to renal artery stenosis or pheochromocytoma (rare tumor of adrenal glands)
Vascular stenosis or aneurysms:
Can lead to strokes or hemorrhage
5. Psychological and Social Impact
Disfigurement from tumors may cause social stigma and low self-esteem
Learning difficulties can impact education and employment
Emotional and behavioral issues may occur
Treatment
1. General Management
Regular monitoring and follow-up with a multidisciplinary team (neurology, dermatology, ophthalmology, orthopedics, genetics)
Early intervention for complications such as learning disabilities, tumors, or skeletal abnormalities
Genetic counseling for affected individuals and families
2. Management of Skin Lesions
Observation: Most cutaneous neurofibromas are benign and don’t require treatment
Surgical removal: For symptomatic, disfiguring, or problematic neurofibromas (painful, bleeding, or impairing function)
Laser therapy: Sometimes used for superficial lesions
3. Treatment of Plexiform Neurofibromas
Surgery: When possible, but complete excision is often difficult due to nerve involvement
Targeted therapies:
MEK inhibitors (e.g., selumetinib): Recently approved for shrinking inoperable plexiform neurofibromas, especially in children
Clinical trials: May be considered for novel therapies
4. Management of Optic Pathway Gliomas
Observation: Many are stable and asymptomatic, especially in young children
Chemotherapy: For progressive tumors causing vision problems (usually carboplatin and vincristine)
Radiation therapy: Used cautiously due to risks in children
5. Skeletal Abnormalities
Orthopedic interventions:
Bracing or surgery for scoliosis
Surgical stabilization for tibial pseudarthrosis
Physical therapy: To maintain mobility and function
6. Neurological and Learning Support
Educational support: For learning disabilities and cognitive challenges
Behavioral therapy: For ADHD or emotional issues
Seizure management: With antiepileptic drugs if needed
7. Management of Hypertension and Vascular Issues
Regular screening for hypertension
Treatment of underlying causes (renal artery stenosis, pheochromocytoma) as appropriate